 砂麼東東
砂麼東東
地中海貧血是一種血紅蛋白異常的疾病,由於常染色體基因缺陷而導致的一種遺傳性疾病。由於調控珠蛋白合成的基因缺失或突變,導致構成血紅蛋白的α鏈和β鏈珠蛋白的合成比例失衡,紅細胞壽命縮短的一種溶血性貧血。
地中海貧血和普通貧血發病機制不同,普通貧血是飲食中缺乏鐵質,導致紅細胞數量過少,紅細胞輸送氧氣不足,而地中海貧血是因紅細胞本身有缺陷,而非紅細胞數量不足。地中海貧血基因攜帶者並不發病,外表和身體機能與常人無異,只有重症患者才會顯露癥狀,治愈可能性較低。
配合世界地中海貧血日(International Thalassaemia Day, ITD),簡稱世界地貧日,設在每年5月8日。本期《空中醫生》邀請到血液腫瘤科專科醫生Dr Leong前來與大家分享地中海貧血症資訊。
1.什麼是地中海貧血?
地中海貧血症,稱之為Thalassemia,是一種遺傳性血液疾病,是因基因突變而導致血紅蛋白(Hemoglobin)異常,進而引發貧血癥狀,而這造成紅血球無法輸送足夠氧氣予身體。地中海貧血症可分類為甲型(Alpha Thalassemia)和乙型(Beta Thalassemia)。
2.地中海貧血患者有哪些明顯癥狀?
其實地中海貧血症的癥狀輕重,是因它的類型不同而有差別,如第一種類型,稱它為攜帶者或帶基因者,通稱為輕型地中海貧血症,而Thalassemia Minor或Thalassemia Trait這類病患,他們沒有明顯癥狀,僅在體檢時被發現有輕微貧血癥狀。
在馬來西亞,約有3至5%人為地中海貧血症的帶基因者,而第二種類型,為中型或重型地中海貧血症,稱之為Thalassemia Intermediate或Thalassemia Major,這類患者有非常明顯癥狀,如心臟和其他器官會因缺氧而衰退,通常是因骨髓無法製造正常的紅血,造成他們患有慢性的嚴重貧血。這類患者一般面色蒼白,容易覺得疲倦,脾臟的脾也會腫大,甚至面對骨骼變形問題,也會有黃疸病狀。
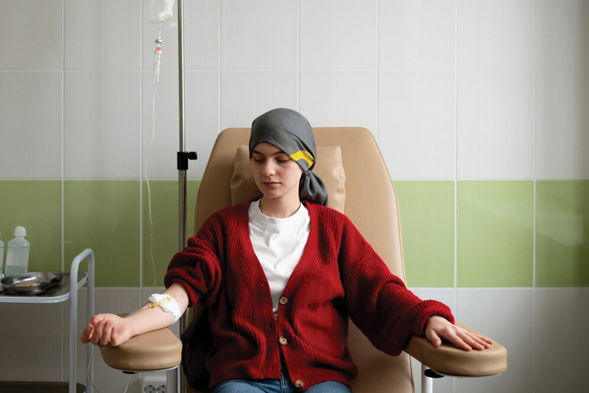
地中海貧血其實也會導致兒童成長髮育遲緩,而重型的地中海貧血症需長期輸血來維持生命,若沒有給予治療,通常會在1至8歲之間逝世。在馬來西亞,約有8千至9千人患有中型或重型地中海貧血症,而在砂拉越,約有200至300名患者屬中型或重型地中海貧血症。
3.如何知道自己是不是地貧基因攜帶者呢?
這可通過數個方式檢測,第一是血液常規檢查,患者可能會出現某些指數偏低如Mcv或Mch會降低,若醫生懷疑患者有地中海貧血症,那他們的血液就會被用血紅蛋白電涌方式(Hemoglobin Electrophoresis)或簡稱為HPLC來分析,這就可以篩查出他們有異常的血紅蛋白。當然最準確但也最昂貴方法是通過基因檢測(Genetic Testing)能更明確判斷一個人是否患有地中海貧血症。
4.如果夫婦雙方都是地貧攜帶者,怎麼辦?
其實我們需知道患者是攜帶者或帶基因者,有關夫婦每胎懷孕會有約25%會誕下重型的地中海貧血症患兒,約50%機率會生出攜帶者患兒,25%機率會有正常胎兒,所以醫生一般會建議在懷孕前接受遺傳疾病檢測。若已經懷孕,可接受產前診斷,如接受羊水穿刺判斷胎兒是否受影響。
文字記錄:楊麗華
資訊出處:Tea FM
官網:teafm.com.my


